皮肤屏障与病毒感染的研究进展
作者:admin 来源:未知 日期:2021-02-04 09:02人气:
摘 要:
病毒感染性皮肤病在人群中具有患病率高、分布广的特点,其发病具有两大关键因素:即屏障破坏与病毒感染宿主细胞。本文就近年来皮肤在抗病毒方面发挥屏障功能以及病毒感染的机制研究进展进行综述。
关键词:
病毒 感染 皮肤屏障
Update of skin barrier and viral infections
LIAN Ni SHI Liqing LIU Lihao CHEN Min
Institute of Dermatology and Hospital of Skin Diseases CAMS & PUMC;
Abstract:
Viral infectious dermatoses are very common in human beings. Host cells infected by virus and skin barrier demage play an important role in the pathogenesis of skin virus infections. The update of the antiviral function of skin barrier and the ways of virus infections is reviewed in this paper.
Keyword:
virus; infection; skin barrier;
病毒在自然界中广泛存在,通过感染宿主细胞实现复制与增殖,并干扰正常的免疫应答,继而引起病理性改变。病毒感染性皮肤病在人群中十分常见。近年来,由于医疗技术和科研水平的逐步提高,人们对病毒感染性皮肤病的发病机制有了更新、更深入的认识。病毒性皮肤病可以粗略分为直接感染皮肤导致的和病毒感染其他系统后的继发皮损(表1)。多数研究仍然认为这种直接感染一般是由于皮肤的破损或微裂隙造成,目前尚未发现病毒可以直接穿过完整、健康的皮肤屏障的依据。
1 皮肤的屏障功能
1.1 物理、化学屏障
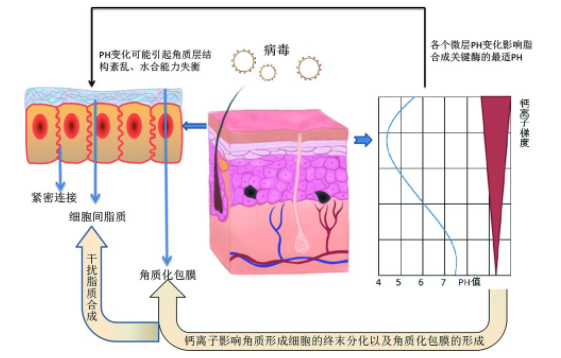
皮肤作为物理屏障的一线屏障功能主要是通过表皮中的角质层实现的[1,2],这层屏障由角质形成细胞、细胞间的脂质和角质桥粒构成。颗粒层上部的角质形成细胞侧壁上的紧密连接也被认为是物理屏障的一部分,被称为二线屏障[3,4]。屏障破坏的原因包括细胞间脂质的分泌减少或成分比例失调、表皮中钙离子梯度的破坏、表皮pH值变化、表皮水合能力失衡以及损伤后的修复能力减退等[5-7]。表皮pH值存在微妙的双相改变,即“S型”梯度,这种模式可以精确地调节表皮各层中脂质合成的关键酶的活性,从而调节脂质合成,并有助于维持角质层中液晶结构的分子顺序。pH值改变同时可能引起细胞间脂质失调、角质层结构改变和水合能力改变,从而进一步导致屏障破坏[7]。过度水合导致角质层厚度增加,但屏障功能减弱,因为角质形成细胞充水肿胀、细胞间隙形成微小的积水,导致角蛋白网格间隙被迫拉大[8]。钙离子梯度破坏将会影响角质形成细胞的终末分化和角质化包膜的形成、阻碍表皮脂质的正常合成以及层状体的胞吐作用等[9]。可以看出,这些因素之间关系密切,相互影响,任何方面的异常都将引起其他功能的改变(图1)。
图1 表皮物理、化学屏障
1.2 免疫屏障
皮肤中免疫细胞在病毒感染早期发挥了“前哨”功能,如角质形成细胞、朗格汉斯细胞、巨噬细胞和树突状细胞等,他们及其分泌的细胞因子限制了病毒从感染部位向周围传播,组成第一道免疫防线[10,11];这些细胞的胞膜表面表达多种模式识别受体(PRR),如:C型凝集素受体、Toll样受体、核苷酸结合寡聚域(NOD)样受体等,可以通过识别部分病毒携带的病原体相关分子模式(PAMP)检测到病毒入侵信号,激活先天免疫[12],并触发下游免疫信号级联反应,成为第二道免疫防线[13,14]。另外,皮肤的“内在免疫”是第三道防线,提供了最直接的抗病毒防御,其介导产生的内在限制因子可以主动结合并直接抑制病毒复制[10,13,15]。
表1 与皮肤病相关的常见病毒
1.3 微生物屏障
皮肤表面定植的共生微生物群可维护皮肤微环境稳态、调节宿主细胞基因的表达,是另一道阻击感染的有力屏障[16,17]。皮肤表面微生物群在维持皮肤稳态和防止微生物突破宿主防御中发挥了重要作用[18-20]。微生物屏障的可能机制:调节性T细胞(Treg)在早期抗原暴露后向皮肤迁移;正常情况下通过Treg产生IL-17以抵御病毒,而在微生物组失衡时,可能会通过皮肤树突状细胞引发Th2轴反应,激活下游炎症因子转录[21,22]。在部分皮肤病的发生发展过程中微生物屏障也扮演了重要角色。有学者研究发现,微生物屏障破坏引起的宿主细胞抗病毒免疫失衡可能激发皮肤炎症,且与IL-23介导的银屑病的发病关系密切[23]。皮肤微生物群、皮肤表面微环境和免疫系统之间相互交叉调节,屏障破坏、菌群失衡可能直接参与了特应性皮炎的发病及反复发作过程[24,25],且由何种微生物群占据主导地位亦与病情轻重有关[26]。另外,病毒也可以存在于正常皮肤表面,最常见的是默克尔多瘤病毒,几乎出现在60%的成人中,但真正发生默克尔细胞瘤的患者却很少[27]。病毒微生物组在皮肤微生物屏障中究竟扮演何种角色尚不明确。
2 病毒感染的机制
2.1 结合与进入
识别宿主细胞受体是病毒感染的初始步骤之一,受体的成功识别可以协调病毒靶向相应组织进行感染并协助其顺利穿越屏障[28]。
病毒感染最常见的细胞受体是唾液酸(sialic acid,SA)受体,多种病毒利用SA受体结合于宿主表皮细胞表面,例如默克尔细胞多瘤病毒,其与SA受体的相互作用可能是感染的初次暴露点,并介导病毒与下游次级蛋白受体发生进一步作用[29]。此外,SA受体可能是决定组织趋向性的关键因素,它可导致菌株特异性和细胞类型依赖性差异,并对病毒性疾病的结局产生重大影响[30]。SA近年来已经成为抗病毒药物研发的新兴靶点[31,32]。部分病毒的识别受体是细胞黏附分子(CAM),许多病毒能够利用CAM潜入狭小密闭的空间,如紧密连接处。最近发现丙型肝炎病毒依赖E-钙黏蛋白作为进入因子,并参与调节上皮-间充质转化[33]。磷脂酰丝氨酸(PtdSer)受体,包括TIM(T细胞免疫球蛋白黏蛋白分子)以及TAM (TYRO3/AXL/MERTK) 酪氨酸激酶受体,被证明是多种包膜病毒的受体[34]。寨卡病毒(ZIKV)通过识别TAM家族成员Tyro 3和AXL感染皮肤成纤维细胞、角质形成细胞和树突状细胞[35]。TIM和TAM同时也是登革热病毒的进入因子[36]。
近期,新型冠状病毒肺炎在世界范围引起大流行,病原体SARS-CoV-2具有高度传染性,引起致命的呼吸道疾病。SARS-CoV-2的刺突蛋白表面包括负责与宿主细胞受体结合的功能亚基(S1)和调控病毒膜与细胞膜的融合的功能亚基(S2)。S1上的受体结合域(receptor-binding domain,RBD)与宿主细胞表面的血管紧张素转化酶2(angiotensin converting enzyme2,ACE2)受体的肽酶结构域(peptidase domain,PD)结合是协助病毒附着的关键步骤,成功识别后,病毒利用蛋白酶跨膜丝氨酸2(TMPRSS2)进一步刺激刺突蛋白,使其在S1/S2和S2’位点裂解,并由S2亚单位驱动病毒膜和宿主细胞膜发生融合[37-39]。
值得注意的是,SARS-CoV-2刺突蛋白的S1/S2交界处新发现的furin裂解位点,是该病毒有别于其他冠状病毒的新特征,若这一裂解位点缺失将对刺突蛋白介导的病毒进入过程产生影响[40,41]。
同时,整合蛋白也可以充当SARS-CoV-2的替代受体,在病毒的黏附、迁移和信号传导过程中发挥作用。学者已经在SARS-CoV-2的刺突蛋白上发现了能与整合蛋白结合的精氨酸-甘氨酸-天冬氨酸(Arg-Gly-Asp, RGD)序列,而其他冠状病毒并不具备这一序列。这段整合蛋白结合序列的位置非常靠近S1-RBD,目前学者们猜测,与整合蛋白的结合可能对ACE2结合起补充作用,如通过整合蛋白发出信号来促进内吞作用,或通过与不同受体结合介导不同的靶细胞感染[42]。
单纯疱疹病毒(HSV)-1通过pH依赖的内吞作用或nectin-1受体依赖的膜融合途径进入表皮角质形成细胞,而通过非pH依赖的病毒包膜与神经元质膜融合的途径进入神经元[43]。另外,HSV-1通过引起肌动蛋白解聚因子(cofilin-1)失活和激活引起肌动蛋白细胞骨架的双相重塑。cofilin-1失活将诱导肌动蛋白聚合,促进病毒进入,而病毒进入后又再次诱导cofilin-1激活,导致聚合的肌动蛋白丝断裂,促进病毒运输[44]。
B组柯萨奇病毒利用DAF(衰变加速因子)介导的信号通路来突破上皮屏障。柯萨奇病毒受体是表皮紧密连接的组成部分,无法从皮肤表面直接接近,因而利用附着在顶端表面的DAF激活Abl激酶,触发Rac依赖的肌动蛋白重排,使病毒得以移动到紧密连接[45]。
2.2 复制与扩散
病毒基因组的复制及其在皮肤中的扩散是成功感染和传播的关键。HSV-1病毒DNA以带有断键缺口和核苷酸空隙的开放式分子形式进入宿主细胞核,启动复制并引起宿主细胞DNA损伤[46]。HSV-1能够破坏宿主细胞的细胞骨架和分泌途径,利用肌动蛋白和微管进行双向转运,即病毒进入时从膜和轴突逆行运输,使其能够沿着感觉轴突将基因组转运至胞核;在病毒装配和退出时则转为顺行运输,再次沿轴突重新扩散至上皮细胞[44]。
ZIKV能够通过诱导皮肤成纤维细胞中的自噬来增强病毒复制[35]。一般地,自噬介导的蛋白降解可以限制病毒复制,但几种虫媒病毒,如登革热病毒、日本脑炎病毒诱导的自噬则通过多种机制清除宿主免疫细胞并促进自身复制和传播[47-49]。因而自噬可能同时具有促病毒作用和抗病毒作用。
水痘-带状疱疹病毒(VZV)感染直接影响角质形成细胞的分化,改变表皮基因表达的正常过程。VZV特异地降低了基底细胞角蛋白和桥粒蛋白的表达,导致表皮结构和功能的破坏,从而重塑表皮环境以促进其自身复制和传播[50]。
既往认为非包膜病毒一般通过胞膜溶解的方式逃脱,但有学者在宿主细胞分泌的细胞外微泡(extracellular microvesicles,EMV)中检测到了病毒核酸,因此猜测EMV提供了一种新的非细胞溶解的扩散方法,这种机制可能会延长病毒复制过程、增强病毒稳定性。因此病毒可能能够 “搭便车”进入病毒通常无法穿透的组织部位,甚至可以绕过血脑屏障进入中枢神经系统[45,51,52]。
2.3 免疫逃逸与免疫调节
宿主抗病毒免疫信号转导途径受多种翻译后修饰的调控,其中包括泛素化。病毒能够利用去泛素化过程欺骗宿主免疫系统,病毒去泛素酶(DUB)与宿主DUB竞争性抑制先天免疫抗病毒信号传导途径,破坏免疫稳态,这种途径可能是病毒在与宿主共同进化时获得了相似机制来逃避免疫监视[53]。
柯萨奇病毒已发展出多种方式来逃避宿主的先天免疫反应。首先,病毒与特定的受体结合,不仅可以协助病毒进入,并且协助其逃避识别;其次,免疫细胞通过识别PAMP和DAMP(损伤相关的分子模式)启动清除病毒的内在化过程,但是病毒同样利用这一过程进行了免疫逃避;另外,通过阻断宿主细胞mRNA的转录和蛋白合成来抑制干扰素相关信号通路、干扰模式识别受体的正常过程以及通过靶向NF-κB信号通路抑制先天免疫应答下游转录因子的激活等[54,55]。
DNA感受器能够感受胞质中游离的病毒DNA并向宿主细胞的固有免疫系统发出潜在的危险信号。cGAS(cyclic GMP-AMP synthase)是一种新型的核酸转移酶,能够感受胞质中游离的病毒DNA,产生内源性环化核苷酸cGAMP并激活干扰素基因刺激(stimulator of interferon genes,STING),从而进一步激活下游干扰素等细胞因子,启动免疫反应。但令人惊讶的是,部分疱疹病毒进化出了一系列针对该信号通路上几乎每个关键步骤的逃逸机制,并利用该途径促进自我复制。首先,HSV-1能够使感受器瘫痪。HSV-1的包膜蛋白VP22能够干扰cGAS正常作用;病毒编码的UL37蛋白是一种脱酰胺酶,可对cGAS产生脱酰胺化作用,从而拮抗cGAMP的产生;病毒编码的UL41蛋白可以介导关闭cGAS/STING介导的胞质DNA传感途径[56-58]。其次,HSV-1可以破坏桥梁分子。HSV-1 VP11/12(由UL46编码)可通过介导STING的降解来阻止STING信号传导[59];HSV-1的γ134.5蛋白通过抑制TANK结合激酶1(TBK1),进而阻断下游干扰素调节因子IRF3/IRF7的活化,抑制干扰素生成,从而促进自身复制和扩散[60]。另外,HSV-1编码多种蛋白可以直接靶向下游转录因子。其中,UL42抑制TNF-α诱导的NF-κB活化并阻止其核易位,而UL24蛋白直接与NF-κB相互作用,抑制其激活易位[61,62]。
登革热病毒基因组编码的七个非结构蛋白(Nonstructural protein,NS蛋白)共同参与免疫逃逸。NS4B与NS5甲基转移酶聚合酶相互作用,使病毒能够逃脱宿主细胞的先天免疫应答;NS3和NS4a阻止视黄酸诱导基因I(RIG-I)易位至线粒体,NS2a和NS4b抑制TBK1的激活,共同阻断RIG-I /线粒体信号通路和β型干扰素(IFN-β)的诱导;NS2b、NS3蛋白酶通过裂解STING编码蛋白来抑制IFN的产生。另外,登革热病毒还可以利用自噬小体进行复制、组装和成熟,并在传播过程中逃避中和抗体[63,64]。ZIKV与登革热病毒非常类似,也是通过包膜蛋白和非结构蛋白(主要是NS1和NS5)操纵宿主细胞,并通过调节干扰素途径和互补拮抗作用来协助病毒免疫逃逸[65]。
有研究发现人疱疹病毒6型(HHV-6)可以特异性抑制NK细胞反应。当感染HHV-6的细胞感受到病毒入侵时,立即增加所有NKG2D(NK细胞活化型受体)配体的mRNA水平以警示免疫系统,而HHV-6则非常迅速的通过抑制NK配体的表达以屏蔽警示系统并下调免疫杀伤[66]。
另外,免疫细胞的功能在很大程度上取决于参与的代谢途径、激活条件和细胞微环境。病毒完全依靠宿主的细胞能量和分子机制进入和复制,并能够模仿、利用、干扰宿主细胞的代谢途径来破坏免疫反应。部分病毒通过靶向线粒体相关蛋白,破坏线粒体膜电位和钙离子平衡、干扰线粒体钙动员、破坏线粒体酶活性以及干扰代谢传感器(例如雷帕霉素复合物1、2机制性靶标mTORC1,mTORC2)来抑制ATP的产生,从而削弱抗病毒免疫应答[67]。
3 小结
皮肤屏障是一种组件复杂、调控精细且一直处在动态变化中的防御系统,各部分能够相互协调,共同抵御病毒入侵。这使得皮肤尽管长期持续地暴露在微生物、病原体下,但严重的皮肤感染并不常发生。本文详细阐述了皮肤屏障应对病毒入侵的防御措施,并对常见病毒感染性皮肤病的病原体突破屏障的策略进行总结。我们发现,病毒能够利用多种策略、从多个方向、在多个水平突破皮肤屏障,并能够抵御、逃避或者调节免疫应答。值得注意的是,尽管已经研究了很多的抗病毒免疫应答的分子机制,但病毒感染后有可能反过来诱导、调节先天性免疫信号,并利用这些信号途径的激活来促进其在宿主中的复制和传播。目前仍有问题待进一步解答:如何提高屏障功能并保护屏障完整性?如何更快更精准的识别病毒入侵信号?如何支持持久的抗病毒免疫应答?病毒具有很强的适应能力,在未来,病毒感染相关性皮肤病的疾病谱可能发生演变。深入研究病毒突破皮肤屏障的机制对未来的临床预防和治疗方面都具有重要意义。
参考文献
[1] Baroni A,Buommino E,De Gregorio V,et al.Structure and function of the epidermis related to barrier properties[J].Clin Dermatol,2012,30(3):257-262.
[2] Wong R,Geyer S,Weninger W,et al.The dynamic anatomy and patterning of skin[J].Exp Dermatol,2016,25(2):92-98.
[3] Ishida-Yamamoto AKM,Honma M.Desmosomes and corneodesmosomes and their relevance to genetic skin diseases[J].G Ital Dermatol Venereol,2017,152(2):148-157.
[4] Jensen JM.PE:The skin's barrier[J].G Ital Dermatol Venereol,2009,144(6):689-700.
[5] Parrish AR.The impact of aging on epithelial barriers[J].Tissue Barriers,2017,5(4):e1343172.
[6] Choi EH.Aging of the skin barrier[J].Clin Dermatol,2019,37(4):336-345.
[7] Wohlrab J,Gebert A,Neubert RHH.Lipids in the skin and pH[J].Curr Probl Dermatol,2018,54:64-70.
[8] Ogawa-Fuse C,Morisaki N,Shima K,et al.Impact of water exposure on skin barrier permeability and ultrastructure[J].Contact Dermatitis,2019,80(4):228-233.
[9] Rinnerthaler M,Streubel MK,Bischof J,et al.Skin aging,gene expression and calcium[J].Exp Gerontol,2015,68:59-65.
[10] Kawamura T,Ogawa Y,Aoki R,et al.Innate and intrinsic antiviral immunity in skin[J].J Dermatol Sci,2014,75(3):159-166.
[11] Kobayashi T,Naik S,Nagao K.Choreographing immunity in the skin epithelial barrier[J].Immunity,2019,50(3):552-565.
[12] Kobayashi T,Ricardo-Gonzalez RR,Moro K.Skin-resident innate lymphoid cells cutaneous innate guardians and regulators[J].Trends Immunol,2020,41(2):100-112.
[13] Nguyen VA,Soulika MA.The dynamics of the skin's immune system[J].Int J Mol Sci,2019.doi:10.3390/ijms20081811
[14] Eyerich S,Eyerich K,Traidl-Hoffmann C,et al.Cutaneous barriers and skin immunity:differentiating a connected network[J].Trends Immunol,2018,39(4):315-327.
[15] Coates M,Blanchard S,MacLeod AS.Innate antimicrobial immunity in the skin:A protective barrier against bacteria,viruses,and fungi[J].PLOS Pathogens,2018,14(12):e1007353.
[16] Belkaid Y,Harrison OJ.Homeostatic immunity and the microbiota[J].Immunity,2017,46(4):562-576.
[17] Meisel JS,Sfyroera G,Bartow-McKenney C,et al.Commensal microbiota modulate gene expression in the skin[J].Microbiome,2018,6(1):20.
[18] Lee E,Lee SY,Kang MJ,et al.Clostridia in the gut and onset of atopic dermatitis via eosinophilic inflammation[J].Ann Allergy Asthma Immunol,2016,117(1):91-92.
[19] Nakamizo S,Egawa G,Honda T,et al.Commensal bacteria and cutaneous immunity[J].Semin Immunopathol,2015,37(1):73-80.
[20] Baldwin HE,Bhatia ND,Friedman A,et al.The Role of Cutaneous Microbiota Harmony in Maintaining a Functional Skin Barrier[J].J Drugs Dermatol,2017,16(1):12-18.
[21] Nowarski R,Jackson R,Flavell RA.The stromal intervention:regulation of immunity and inflammation at the epithelial-mesenchymal barrier[J].Cell,2017,168(3):362-375.
[22] Linehan JL,Harrison OJ,Han SJ,et al.Non-classical immunity controls microbiota impact on skin immunity and tissue repair[J].Cell,2018,172(4):784-796.
[23] Zhu H,Lou F,Yin Q,et al.RIG-I antiviral signaling drives interleukin-23 production and psoriasis-like skin disease[J].EMBO Mol Med,2017,9(5):589-604.
[24] Kim BE,Leung DYM.Significance of skin barrier dysfunction in atopic dermatitis[J].Allergy Asthma Immunol Res,2018,10(3):207-215.
[25] Chng KR,Tay ASL,Li C,et al.Whole metagenome profiling reveals skin microbiome-dependent susceptibility to atopic dermatitis flare[J].Nat Microbiol,2016,1(9):16106.
[26] Byrd AL,Deming C,Cassidy SKB,et al.Staphylococcus aureus and Staphylococcus epidermidis strain diversity underlying pediatric atopic dermatitis[J].Sci Transl Med,2017,9(397):eaal4651.
[27] Moore PS,Chang Y.Common commensal cancer viruses[J].PLOS Pathogens,2017,13(1):e1006078.
[28] Maginnis MS.Virus-receptor interactions:the key to cellular invasion[J].J Mol Biol,2018,430(17):2590-2611.
[29] Stehle T,Khan ZM.Rules and exceptions:sialic acid variants and their tole in determining viral tropism[J].J Virol,2014,88(14):7696.
[30] Shi Y,Wu Y,Zhang W,et al.Enabling the ‘host jump’:structural determinants of receptor-binding specificity in influenza A viruses[J].Nat Rev Microbiol,2014,12(12):822-831.
[31] Govorkova EA,Baranovich T,Marathe BM,et al.Sialic acid-binding protein Sp2CBMTD protects mice against Lethal challenge with emerging influenza a (H7N9) virus[J].Antimicrob Agents Chemother,2015,59(3):1495.
[32] Mayberry CL,Nelson CDS,Maginnis MS.JC Polyomavirus Attachment and Entry:Potential Sites for PML Therapeutics[J].Curr Clin Microbiol Rep,2017,4(3):132-141.
[33] Li Q,Sodroski C,Lowey B,et al.Hepatitis C virus depends on E-cadherin as an entry factor and regulates its expression in epithelial-to-mesenchymal transition[J].Proceedings of the National Academy of Sciences,2016,113(27):7620.
[34] Moller-Tank S,Maury W.Phosphatidylserine receptors:Enhancers of enveloped virus entry and infection[J].Virology,2014,11:468-470,565-580.
[35] Hamel R,Dejarnac O,Wichit S,et al.Biology of zika virus infection in human skin cells[J].J Virol,2015,89(17):8880-8896.
[36] Meertens L,Carnec X,Lecoin Manuel P,et al.The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry[J].Cell Host & Microbe,2012,12(4):544-557.
[37] Hoffmann M,Kleine-Weber H,Schroeder S,et al.SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor[J].Cell,2020,181(2):271-280.
[38] Wang Q,Zhang Y,Wu L,et al.Structural and functional basis of SARS-CoV-2 entry by using human ACE2[J].Cell,2020.doi:https://doi.org/10.1016/j.cell.2020.03.045.
[39] Lan J,Ge J,Yu J,et al.Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor[J].Nature,2020.doi:10.1038/s41586-020-2180-5.
[40] Yan R,Zhang Y,Li Y,et al.Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2[J].Science,2020,367(6485):1444.
[41] Walls AC,Park YJ,Tortorici MA,et al.Structure,function,and antigenicity of the SARS-CoV-2 spike glycoprotein[J].Cell,2020,181(2):281-292.
[42] Sigrist CJA,Bridge A,Le Mercier P.A potential role for integrins in host cell entry by SARS-CoV-2[J].Antiviral Res,2020,177:104759.
[43] Sayers CL,Elliott G.Herpes simplex virus 1 enters human keratinocytes by a nectin-1-dependent,rapid plasma membrane fusion pathway that functions at low temperature[J].J Virol,2016,90(22):10379-10389.
[44] Miranda-Saksena M,Denes EC,Diefenbach JR,et al.Infection and transport of herpes simplex virus type 1 in neurons:role of the cytoskeleton[J].Viruses,2018.doi:10.3390/v10020092.
[45] Sin J,Mangale V,Thienphrapa W,et al.Recent progress in understanding coxsackievirus replication,dissemination,and pathogenesis[J].Virology,2015,484:288-304.
[46] Kobiler O,Weitzman MD.Herpes simplex virus replication compartments:From naked release to recombining together[J].PLOS Pathogens,2019,15(6):e1007714.
[47] Li JK,Liang JJ,Liao CL,et al.Autophagy is involved in the early step of Japanese encephalitis virus infection[J].Microbes Infection,2012,14(2):159-168.
[48] Heaton NS,Randall G.Dengue virus and autophagy[J].Viruses,2011.doi:103390/v3081332
[49] Corona AK,Saulsbery HM,Corona Velazquez AF,et al.Enteroviruses remodel autophagic trafficking through regulation of host SNARE proteins to promote virus replication and cell exit[J].Cell Reports,2018,22(12):3304-3314.
[50] Jones M,Dry IR,Frampton D,et al.RNA-seq analysis of host and viral gene expression highlights interaction between varicella zoster virus and keratinocyte differentiation[J].PLOS Pathogens,2014,10(1):e1003896.
[51] Robinson SM,Tsueng G,Sin J,et al.Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers[J].PLOS Pathogens,2014,10(4):e1004045.
[52] Sampey GC,Meyering SS,Asad Zadeh M,et al.Exosomes and their role in CNS viral infections[J].J Neuro Virology,2014,20(3):199-208.
[53] Kumari P,Kumar H.Viral deubiquitinases:role in evasion of anti-viral innate immunity[J].Crit Rev Microbiol,2018,44(3):304-317.
[54] Zhang Y,Li J,Li Q.Immune evasion of enteroviruses vnder innate immune monitoring[J].Front Microbiol,2018,9:1866.
[55] Lei X,Xiao X,Wang J.Innate immunity evasion by enteroviruses:insights into virus-host interaction[J].Viruses,2016.doi:10.3390/v8010022.
[56] Huang JYH,Su C,Li Y,et al.Herpes simplex virus 1 tegument protein VP22 abrogates cGAS/STING-mediated antiviral innate immunity[J].J Virol,2018.doi:10.1128/JVI.00841-18.
[57] Su C,Zheng C.Herpes simplex virus 1 abrogates the cGAS/STING-Mediated cytosolic DNA-sensing pathway via its virion host shutoff protein,UL41[J].J Virol,2017.doi:10.1128/JVI.02414-16.
[58] Zhang J,Zhao J,Xu S,et al.Species-specific deamidation of cGAS by herpes simplex virus UL37 protein facilitates viral replication[J].Cell Host Microbe,2018,24(2):234-248.
[59] Deschamps TKM.Evasion of the STING DNA-sensing pathway by VP11/12 of herpes simplex virus 1[J].J Virol,2017.doi:10.1128/JVI.00535-17.
[60] Richard Manivanh JM,David M,et al.Leib:Role of herpes simplex virus 1 γ34.5 in the regulation of IRF3 signaling[J].J Virol,2017.doi:10.1128/JVI.01156-17.
[61] Zhang J,Wang S,Wang K,et al.Herpes simplex virus 1 DNA polymerase processivity factor UL42 inhibits TNF-α-induced NF-κB activation by interacting with p65/RelA and p50/NF-κB1[J].Med Microbiol Immunol,2013,202(4):313-325.
[62] Xu H,Su C,Pearson A,et al.Herpes simplex virus 1 UL24 abrogates the DNA sensing signal pathway by inhibiting NF-kappaB activation[J].J Virol,2017.doi:10.1128/JVI.00025-17.
[63] Lescar J,Soh S,Lee LT,et al.The dengue virus replication complex:from RNA replication to protein-protein interactions to evasion of innate immunity.in:dengue and zika:control and antiviral treatment strategies.edn.edited by hilgenfeld R,Vasudevan SG[M].Singapore:Springer Singapore,2018:115-129.
[64] Uno N,Ross TM.Dengue virus and the host innate immune response[J].Emerging Microbes & Infections,2018,7(1):1-11.
[65] Asif A,Manzoor S,Tuz-Zahra F,et al.Zika virus:immune evasion mechanisms,currently available therapeutic regimens,and vaccines[J].Viral Immunol,2017,30(10):682-690.
[66] Schmiedel D,Tai J,Levi-Schaffer F,et al.Human herpesvirus 6B downregulates expression of activating ligands during lytic infection to escape elimination by natural killer cells[J].J Virol,2016,90(21):9608-9617.
[67] Moreno-Altamirano MMB,Kolstoe SE,Sanchez-Garcia FJ.Virus control of cell metabolism for replication and evasion of host immune responses[J].Front Cell Infect Microbiol,2019,9:95.

